Does TAMOF Exist? Revisiting a Diagnosis I Thought I Understood
- Mar 5
- 6 min read
Earlier this year, I wrote a piece about TAMOF — thrombocytopenia-associated multiple organ failure — and the case for recognizing it as a TTP-like process driven by secondary ADAMTS13 deficiency. I described the lab pattern. I walked through the differential. I made what I believed was a compelling argument that TAMOF is underrecognized, and that plasma exchange is a rational intervention once the diagnosis is made.
I stand by that piece. The pathophysiology is real. The lab pattern is real. The clinical scenarios I described are ones that pathologists and intensivists encounter regularly.
But since writing it, I’ve spent more time with the literature surrounding TAMOF, and I’ve come to appreciate something I didn’t fully reckon with the first time around: the question isn’t just whether TAMOF is real. The question is whether TAMOF is useful — and that turns out to be a much harder thing to answer.
The Case I Made
In my earlier article, I described TAMOF as occupying an uncomfortable space between DIC, TTP, and sepsis-associated coagulopathy. The central idea was that systemic inflammation can drive a relative deficiency of ADAMTS13, leading to accumulation of ultra-large von Willebrand factor multimers and platelet-rich microthrombi in the microvasculature. The downstream effect looks like TTP — organ ischemia, thrombocytopenia, elevated LDH, sometimes schistocytes — even though the trigger is sepsis or inflammation rather than autoimmunity.
I emphasized that the lab pattern tells the story: falling platelets, rising LDH, preserved coagulation parameters, and organ dysfunction out of proportion to hemodynamics. I argued that recognizing this pattern opens the door to therapeutic plasma exchange, and that missing it leaves patients undertreated.
None of that is wrong, exactly. But it is incomplete.
The Questions I Didn’t Ask
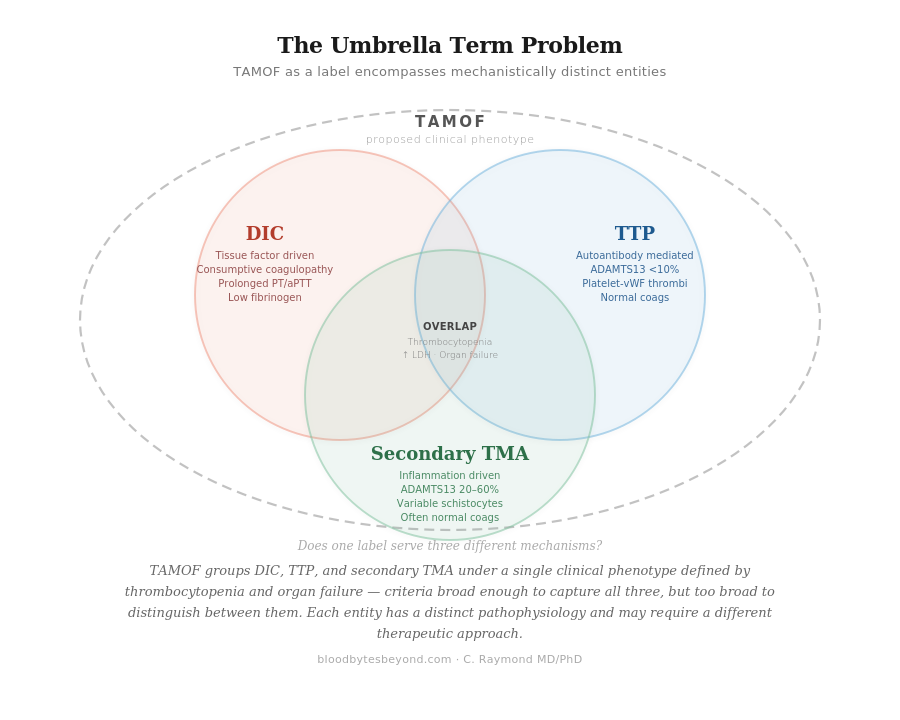
The concept of TAMOF was primarily developed by Nguyen and Carcillo, with foundational work published in Critical Care in 2006. From the beginning, TAMOF was described not as a single disease but as a clinical phenotype — an umbrella encompassing TTP, DIC, and secondary thrombotic microangiopathy in critically ill patients. The unifying feature was new-onset thrombocytopenia coinciding with multiple organ failure, and the proposed mechanism was microvascular thrombosis.
This is where the first tension appears. If TAMOF includes DIC, TTP, and secondary TMA under one label, what does the label add? Each of those entities already has its own diagnostic criteria, its own pathophysiology, and — critically — its own treatment approach. DIC is a consumptive coagulopathy driven by tissue factor. TTP is an autoantibody-mediated deficiency of ADAMTS13. Secondary TMA is a broader category of inflammation-driven microangiopathy. These are not the same process. Grouping them under one name risks implying a mechanistic unity that does not exist.
In my first article, I focused on the subset of TAMOF that behaves like TTP — the secondary TMA piece, where inflammation drives ADAMTS13 deficiency and platelet-vWF-mediated thrombosis predominates. That is a real phenomenon. But by calling it TAMOF rather than secondary TMA, I may have inadvertently adopted a framework that obscures more than it clarifies.
The Evidence Problem
The therapeutic implication of recognizing TAMOF is plasma exchange. This was central to my earlier piece: once you see the microangiopathy, the rationale for plasma exchange follows logically. Remove the ultra-large vWF multimers. Replenish ADAMTS13. Reduce inflammatory mediators.
The biological plausibility is sound. The evidence base, however, is thin.
The landmark pediatric trial randomized just ten children — five to plasma exchange, five to standard therapy. The results were encouraging: plasma exchange restored ADAMTS13 activity and was associated with organ failure resolution. But a trial of ten patients, however well-designed, cannot establish standard of care. Subsequent studies have been retrospective, observational, or limited to small cohorts. The Turkish TAMOF Network described outcomes in 42 children but could not even measure ADAMTS13 levels due to unavailability of the assay. A prospective multicenter experience published more recently found lower 28-day mortality in children treated with plasma exchange, but the authors themselves concluded that a randomized clinical trial is necessary to establish a causal relationship.
The American Society for Apheresis gives plasma exchange in sepsis with multiple organ failure a Category III recommendation — meaning the optimum role is not established and decision-making should be individualized. This is not an endorsement. It is an acknowledgment that we don’t know enough.
The ADAMTS13 Problem
In my earlier piece, I described ADAMTS13 as spanning a wide range in TAMOF and cautioned against rigid thresholds. I still think that’s right. But I underappreciated a more fundamental issue: we do not yet know whether reduced ADAMTS13 in sepsis is the cause of organ dysfunction or simply a marker of disease severity.
This distinction matters enormously. If reduced ADAMTS13 is pathogenic — if it is actively driving microthrombi formation and organ ischemia — then replenishing it through plasma exchange is a targeted intervention. But if ADAMTS13 is reduced because the patient is severely ill, because inflammation broadly suppresses hepatic synthesis and accelerates consumption of many proteins, then treating the ADAMTS13 level may be treating a surrogate rather than the disease itself.
Moreover, ADAMTS13 activity in TAMOF is typically reduced but not severely deficient. In classic TTP, levels are usually below 10%. In sepsis-associated secondary TMA, levels are more often in the 20–60% range. This is an important gray zone. Plenty of critically ill patients with sepsis have mildly reduced ADAMTS13, and most of them do not have a clinically meaningful microangiopathic process. The specificity of this biomarker, in this context, is genuinely uncertain.
The Diagnostic Boundary Problem
TAMOF is diagnosed by a triad: new-onset thrombocytopenia below 100,000, at least two failing organs, and elevated LDH. The problem is that this triad describes an enormous proportion of critically ill septic patients. Thrombocytopenia in the ICU is common — present in up to 40–50% of patients, depending on the threshold used. LDH elevation is nearly ubiquitous in critical illness. And organ failure is, almost by definition, why these patients are in the ICU in the first place.
If the diagnostic criteria capture too many patients, the label loses its power to identify those who would specifically benefit from targeted intervention. A diagnosis that applies to half the ICU is not a diagnosis. It is a description.
What I Think Now
I want to be careful here, because I don’t think the answer is that TAMOF is meaningless or that my earlier article was misguided. The pathophysiology I described — inflammation-driven ADAMTS13 deficiency leading to platelet-vWF-mediated microvascular thrombosis — is supported by autopsy data, by biomarker studies, and by the clinical observation that some septic patients develop a microangiopathic picture that does not fit neatly into DIC. That phenomenon is real, and it deserves a name.
But I think the name might be doing some work that the evidence hasn’t earned yet.
TAMOF as an umbrella term bundles together mechanistically distinct processes and implies they share a common therapeutic target. TAMOF as a diagnostic entity relies on criteria so broad that they risk capturing patients who don’t have a true microangiopathy at all. And TAMOF as a justification for plasma exchange rests on studies that, while promising, remain small, largely retrospective, and without a definitive randomized trial.
What I wrote before was an argument for recognition — for seeing the pattern and acting on it. What I’d add now is an argument for precision. The lab pattern I described is still the right place to start. Converging signals of microangiopathy in a septic patient should prompt the question: is there a thrombotic microangiopathic process driving this patient’s organ failure? But the answer to that question should lead to a specific diagnosis — secondary TMA, DIC, or something else — not to a catch-all label that may prematurely close the differential.
The Lab’s Role, Revisited
I ended my first article with the line: “TAMOF is not rare because it is uncommon. It is rare because we don’t look for it.”
I still believe that’s true — but I’d frame it differently now. What’s underrecognized isn’t necessarily TAMOF as a discrete entity. What’s underrecognized is the broader phenomenon of secondary thrombotic microangiopathy in the critically ill, and the role that laboratory medicine plays in distinguishing it from DIC, from “just sepsis,” and from true TTP. That distinction requires more than a label. It requires the kind of contextual interpretation that has always been the core competency of the pathologist: not just reporting numbers, but assembling them into a story that changes management.
The controversy around TAMOF is not really about whether the biology is real. It is about whether we have the right framework to describe it, the right criteria to diagnose it, and the right evidence to treat it. On all three counts, the honest answer is: not yet.
But “not yet” is not the same as “no.”
It means we have more work to do. And for those of us in the lab, that work starts with being willing to question our own frameworks — even the ones we just finished building.



